What Is Solid-Phase Peptide Synthesis? A Research Overview
Solid-phase peptide synthesis (SPPS) is the method by which the vast majority of research-grade synthetic peptides are produced today. Developed in the early 1960s and refined over subsequent decades, SPPS transformed peptide science from a painstaking manual discipline into a precise, reproducible, and scalable laboratory process. For researchers working with synthetic peptide compounds, understanding how SPPS works — and why it matters for compound quality — provides essential context for evaluating the peptides used in experimental settings. This overview covers the principles, process steps, key chemistry variants, and research relevance of solid-phase peptide synthesis.
The Origins of Solid-Phase Peptide Synthesis
Before SPPS existed, synthesizing a peptide in the laboratory was an extraordinarily labour-intensive undertaking. Each amino acid addition required its own set of chemical reactions, purification steps, and yield assessments. A peptide of even modest length — ten to fifteen residues — could take months of skilled laboratory work to produce, with significant losses of material at each stage.
The breakthrough came in 1963 when American biochemist Bruce Merrifield introduced a fundamentally different approach. Rather than performing synthesis in solution — where each intermediate had to be isolated and purified — Merrifield proposed anchoring the first amino acid to an insoluble solid support, commonly referred to as a resin. Subsequent amino acids could then be added in a stepwise fashion, with excess reagents and byproducts washed away between each coupling step without the need to isolate intermediate products.
The elegance of this approach lay in its simplicity: the growing peptide chain stayed fixed to the resin throughout the entire synthesis, and only at the end was the finished peptide cleaved free and purified. Merrifield was awarded the Nobel Prize in Chemistry in 1984 for this contribution, which remains the dominant method for producing synthetic research peptides to this day.
Core Principles of the SPPS Method
The SPPS method operates on a repeating cycle of chemical steps, each performed while the growing peptide chain remains anchored to the solid resin support. The key principles that make this system work are:
- Solid support anchoring: The C-terminal amino acid of the target sequence is covalently attached to the resin at the start of synthesis. This anchor point holds the chain throughout all subsequent steps.
- Protecting group chemistry: Amino acids carry protecting groups on their reactive functional groups to prevent unwanted side reactions during coupling. These groups are selectively removed at the appropriate stage of synthesis.
- Stepwise elongation: Amino acids are added one at a time from the C-terminus to the N-terminus — the opposite direction to natural ribosomal synthesis — in a precise sequence determined by the target peptide.
- Wash and cycle: After each coupling step, the resin is washed thoroughly to remove unreacted reagents before the next amino acid is introduced. This eliminates the need for intermediate purifications.
- Final cleavage and purification: Once the full sequence has been assembled, the completed peptide is cleaved from the resin and the protecting groups are removed simultaneously, yielding the crude peptide which is then purified — typically by HPLC — to research-grade purity.
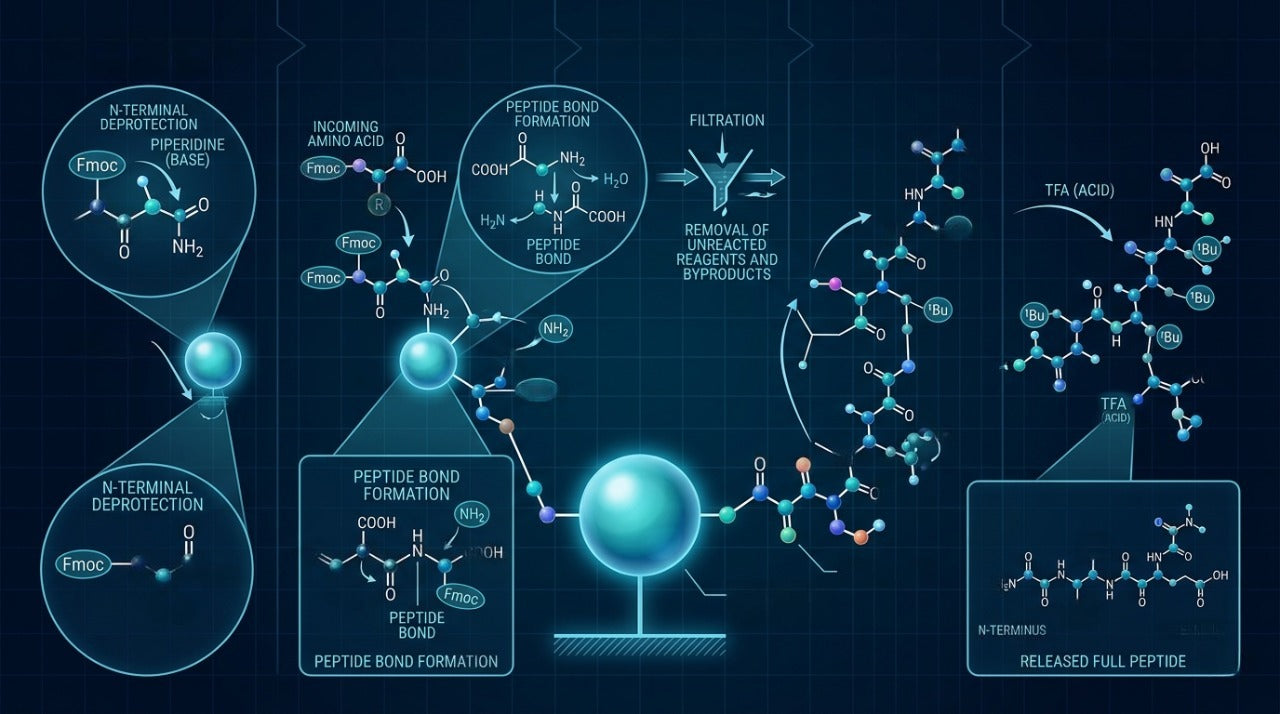
Step-by-Step: How SPPS Works in Practice
A standard SPPS cycle proceeds through the following stages for each amino acid addition:
- Deprotection: The protecting group on the N-terminus of the resin-bound chain is removed, exposing the free amino group ready for the next coupling reaction. In Fmoc SPPS (see below), this is achieved using a piperidine solution.
- Washing: The resin is washed multiple times with solvent to remove all traces of the deprotection reagent before coupling begins.
- Coupling: The next protected amino acid — activated at its carboxyl group using a coupling reagent — is introduced and reacts with the free amino group on the resin-bound chain, forming a new peptide bond.
- Capping (optional): Any unreacted amino groups are chemically capped to prevent them from participating in future coupling steps, which would produce truncated sequences in the final product.
- Washing: The resin is washed again to remove excess activated amino acid and coupling reagents before the cycle repeats.
This cycle repeats for every residue in the target sequence. For a 20-residue peptide, the cycle runs twenty times. Modern automated synthesizers execute each cycle with computer-controlled precision, monitoring reagent volumes, reaction times, and wash steps with minimal human intervention.
Fmoc vs. Boc Chemistry: The Two Main SPPS Strategies
Two principal protecting group strategies are used in modern solid-phase peptide synthesis, each with distinct advantages for different research applications:
Fmoc (9-Fluorenylmethyloxycarbonyl) Chemistry
Fmoc chemistry uses a base-labile protecting group that is removed under mild conditions — typically a 20% piperidine solution in DMF. Because deprotection does not require strong acid, Fmoc SPPS is compatible with a wider range of sensitive amino acid side chains and resin types. It is the most widely used strategy in modern research peptide production, favoured for its compatibility with automated synthesizers and its relatively mild reaction conditions.
Boc (tert-Butyloxycarbonyl) Chemistry
Boc chemistry uses an acid-labile protecting group removed by trifluoroacetic acid (TFA) treatment at each cycle. Final cleavage from the resin requires hydrofluoric acid (HF), a highly corrosive reagent that demands specialized laboratory equipment and handling protocols. Despite these demands, Boc chemistry offers advantages for certain difficult sequences and remains in use for specific research applications where Fmoc chemistry performs poorly.
For most research peptide production purposes, Fmoc chemistry is the default choice due to its practical accessibility and broad compatibility.
Quality Outcomes: What SPPS Means for Research Compound Purity
The quality of a synthetic peptide produced by SPPS is determined by the precision with which each cycle is executed and the thoroughness of the final purification step. Several factors influence the purity of the finished research compound:
- Coupling efficiency: If a coupling step is incomplete, a fraction of chains will lack one residue — producing deletion sequences that co-purify with the target peptide if not adequately resolved by HPLC
- Side chain protection: Incomplete removal of protecting groups during cleavage leaves modified residues in the final product, reducing purity and potentially altering the compound's behaviour in assays
- Racemization: Under suboptimal coupling conditions, amino acid residues can undergo racemization — converting from their natural L-form to the D-form — introducing structural impurities that may affect experimental reproducibility
- HPLC purification: Reverse-phase HPLC is the standard final purification step for research peptides, resolving the target sequence from truncated sequences, deletion products, and protecting group remnants
Research-grade peptides produced by rigorous SPPS and purified by preparative HPLC are typically characterized at ≥98% purity by analytical HPLC, with identity confirmed by mass spectrometry. These standards are what distinguish research-grade compounds from lower-quality synthetic products.
Frequently Asked Questions
What does solid-phase mean in solid-phase peptide synthesis?
The term solid-phase refers to the insoluble resin support to which the growing peptide chain is anchored throughout the synthesis process. Unlike solution-phase synthesis — where reactions occur between dissolved molecules and intermediates must be isolated at each step — solid-phase synthesis keeps the peptide chain fixed in place, allowing reagents to be added and washed away without isolating intermediate products. This is the core innovation that makes SPPS so practical and scalable compared to earlier methods.
How long does it take to synthesize a research peptide using SPPS?
Synthesis time depends primarily on the length of the target sequence and whether synthesis is performed manually or on an automated platform. Modern automated synthesizers can typically complete the coupling cycles for a 10 to 20 residue peptide within a few hours. Longer or more complex sequences may require additional time. Following synthesis, cleavage, and purification by HPLC add further time — most research peptides are ready for quality analysis within one to several days of synthesis initiation, depending on laboratory workflow.
Why is HPLC purification necessary after SPPS?
No synthesis cycle achieves 100% coupling efficiency. Even small percentages of incomplete couplings at each step accumulate across a multi-residue sequence, producing a crude product that contains the target peptide alongside truncated sequences, deletion products, and residual protecting group fragments. Reverse-phase HPLC separates these impurities from the target compound based on differences in hydrophobicity, yielding a purified product that meets research-grade purity standards. Without this step, the compound's identity and purity cannot be reliably verified.
Is solid-phase peptide synthesis used for all research peptides?
SPPS is the dominant method for producing synthetic research peptides up to approximately 50 residues in length. For longer sequences, researchers may use alternative strategies such as native chemical ligation — a technique that joins two shorter SPPS-produced peptide fragments together. Very large peptide sequences or proteins may be produced through recombinant expression systems rather than chemical synthesis. For the research peptide compounds most commonly used in laboratory settings, however, SPPS remains the standard production method.
Research Purposes Disclaimer
All content in this article is provided for educational and scientific research purposes only. The information presented is based on publicly available scientific literature and is intended to inform researchers and science-interested readers about peptide synthesis methodology. Nothing in this article constitutes medical advice, clinical guidance, or a recommendation for human or animal consumption of any compound. Peptide research compounds are laboratory reagents intended solely for use in controlled scientific research environments and are not approved for human consumption, therapeutic use, or diagnostic application. Researchers are responsible for ensuring full compliance with all applicable institutional, regional, and national regulations governing the procurement, handling, and use of research compounds.